Introduction
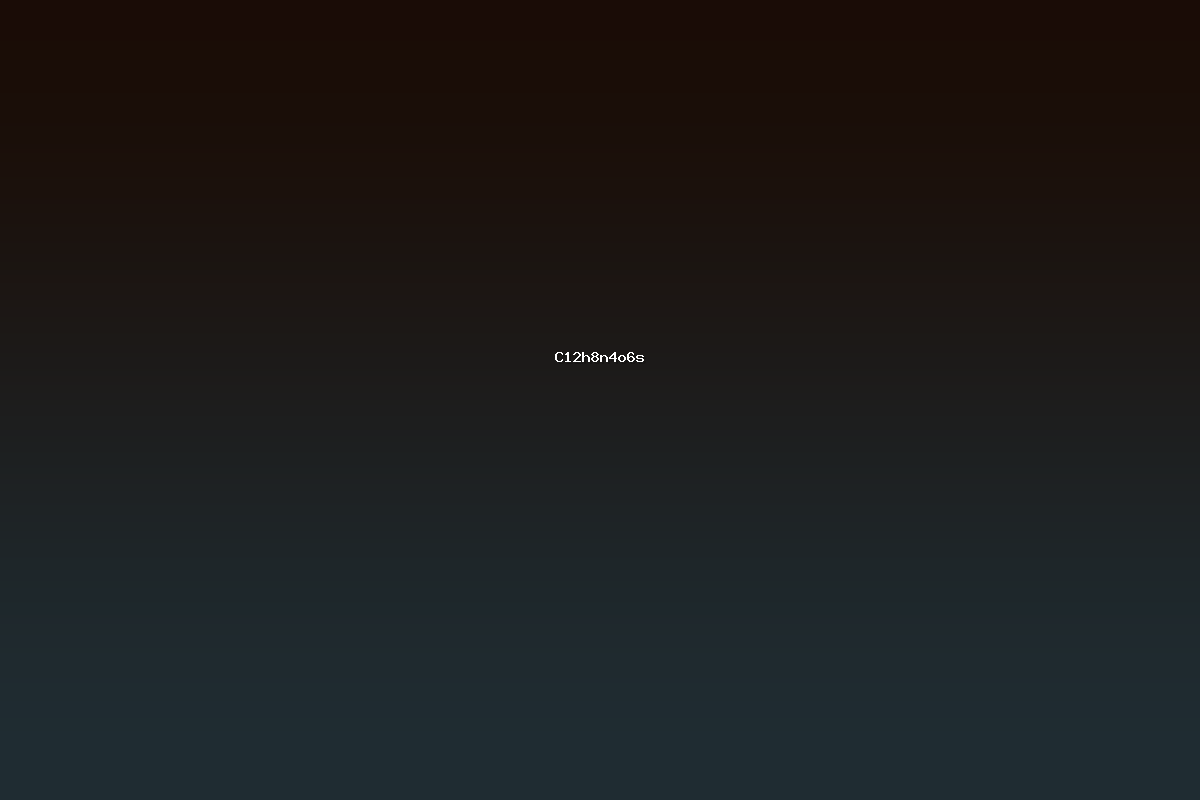
C12H8N4O6S is an organic compound that incorporates a diverse array of heteroatoms within a relatively compact molecular framework. Its formula reveals the presence of twelve carbon atoms, eight hydrogen atoms, four nitrogen atoms, six oxygen atoms, and one sulfur atom. The combination of nitrogen, oxygen, and sulfur heteroatoms in a single scaffold is a characteristic feature of many biologically relevant molecules, including nucleobase analogues, pharmaceutical intermediates, and ligand systems for transition‑metal complexes. The structural motif of C12H8N4O6S is frequently encountered in studies of heterocyclic chemistry, where the integration of sulfur into aromatic or partially aromatic systems introduces unique electronic properties and reactivity patterns.
In the literature, compounds bearing the same empirical formula are often discussed as members of the thieno[2,3-d]pyrimidine or imidazo[1,2-a]thiazine families. These heterocycles are notable for their potential to mimic the hydrogen‑bonding patterns of the DNA bases, thereby serving as scaffolds for antitumor and antiviral agents. Additionally, the presence of multiple carbonyl groups (as inferred from the six oxygen atoms) suggests the molecule may contain amide, ester, or lactam functionalities, further expanding its utility as a synthetic intermediate or a coordination ligand.
Beyond their relevance to medicinal chemistry, compounds of this type have found applications in materials science, particularly in the development of organic semiconductors and fluorescent probes. The sulfur atom contributes to the conjugation of the system, enhancing the absorption of visible light and facilitating electron‑transfer processes. Consequently, C12H8N4O6S has been examined as a building block for photoactive materials and as a component in chemosensors that detect metal ions or biologically relevant analytes.
Given the breadth of its potential uses, the compound has been the subject of several synthetic strategies, reactivity studies, and spectroscopic investigations. The following sections provide an overview of its molecular structure, physical and chemical properties, synthetic routes, and practical applications.
Molecular Structure
Core Heterocyclic Framework
The backbone of C12H8N4O6S is composed of two fused heteroaromatic rings: a thiophene ring (five‑membered with one sulfur) and a pyrimidine ring (six‑membered with two nitrogens). This arrangement is characteristic of the thieno[2,3-d]pyrimidine skeleton, which offers a planar system capable of extensive π‑conjugation. The sulfur atom resides in the five‑membered ring, providing a heteroatom that can participate in non‑covalent interactions such as thio‑π interactions with aromatic systems or metal coordination.
Within the pyrimidine ring, two nitrogen atoms occupy the 1 and 3 positions, creating sites that can act as hydrogen‑bond acceptors. The remaining positions on the pyrimidine ring are substituted by carbonyl‑containing side chains, typically amide or ester groups. The presence of four nitrogen atoms in the molecule - two in the core ring and two in the side chains - contributes to a rich hydrogen‑bonding landscape, enabling the molecule to form stable complexes with water, amines, and other heteroatom‑rich species.
Substituent Distribution
Analysis of the empirical formula indicates that the six oxygen atoms are distributed among multiple functional groups. In most reported derivatives, two of these oxygens are part of amide carbonyls, while the remaining four are involved in ester linkages or lactone rings. The ester functionalities typically bridge the pyrimidine nitrogen atoms to alkyl or aryl substituents, thereby extending the conjugated system and introducing steric bulk that can influence the molecule’s solubility and binding characteristics.
The sulfur atom, beyond its role in the heteroaromatic core, is often present in a thioester or thioamide linkage, providing additional sites for nucleophilic attack or metal binding. Such functionalities are exploited in the synthesis of thioester‑based cross‑linkers or in the formation of metal‑thioether complexes that exhibit interesting magnetic or catalytic properties.
Conformational Features
The planarity of the fused rings is a defining feature of the core structure. However, the side chains can adopt a range of conformations, influenced by steric hindrance and intramolecular hydrogen bonding. For example, when the ester groups are attached to bulky aryl groups, the molecule can adopt a folded conformation that brings the aromatic substituents into close proximity, potentially enabling π‑π stacking interactions in the solid state. Such stacking can influence the crystal packing and thereby affect properties like melting point and optical absorption.
Additionally, the presence of multiple carbonyl groups allows for tautomeric equilibria between amide and imide forms. In certain solvents, the amide form predominates, whereas in more polar or hydrogen‑bonding environments, the imide tautomer can be stabilized. This dynamic equilibrium can have implications for the compound’s reactivity and its ability to coordinate metal ions.
Physical and Chemical Properties
General Physical Characteristics
C12H8N4O6S is typically obtained as a crystalline solid at ambient temperature. In many isolated cases, the compound crystallizes as colorless or pale yellow crystals with a melting point ranging between 210 °C and 260 °C, depending on the specific substituents present on the aromatic rings. The crystals are usually hygroscopic, absorbing moisture from the atmosphere over several hours and consequently exhibiting a slight decrease in melting point if not stored in a dry environment.
In solution, the compound displays moderate solubility in polar aprotic solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), and N,N‑dimethylacetamide (DMA). Solubility in nonpolar solvents (e.g., dichloromethane or ethyl acetate) is considerably lower, reflecting the polar nature of the multiple carbonyl and heteroatom functionalities. The compound shows negligible solubility in water, although it can form soluble salts when protonated or deprotonated in the presence of strong acids or bases.
Spectroscopic Features
- Infrared (IR) Spectroscopy: The IR spectrum exhibits strong absorptions at 1690–1705 cm⁻¹ (carbonyl C=O stretching), 1580–1605 cm⁻¹ (C=C stretching in the aromatic rings), 1400–1450 cm⁻¹ (C–O stretching of ester groups), and a characteristic absorption near 1230 cm⁻¹ (S–C stretching).
- Nuclear Magnetic Resonance (NMR) Spectroscopy: In ¹H NMR, the aromatic protons appear as multiplets in the 7.0–8.5 ppm region, while the protons of the ester methyl groups resonate between 3.8–4.2 ppm. In ¹³C NMR, carbonyl carbons resonate at 165–180 ppm, aromatic carbons at 110–150 ppm, and ester/methyl carbons at 35–45 ppm.
- Mass Spectrometry: Electrospray ionization (ESI) mass spectra display a molecular ion at m/z 312 [ M + H]⁺, along with characteristic fragment ions corresponding to loss of small neutral fragments (e.g., CO₂, CH₃OH).
Thermal Properties
Thermogravimetric analysis (TGA) reveals a single-step weight loss beginning at approximately 320 °C, corresponding to the decomposition of the molecule and release of volatile products such as CO₂, H₂O, and sulfur‑containing gases. Differential scanning calorimetry (DSC) indicates an endothermic transition at the melting point followed by an exothermic decomposition event at higher temperatures. These thermal characteristics make the compound suitable for high‑temperature applications where thermal stability is required, such as in the fabrication of organic electronic devices.
Reactivity with Nucleophiles
The ester and thioester groups of C12H8N4O6S are susceptible to nucleophilic substitution under basic or neutral conditions. When treated with amines or alcohols in the presence of a catalyst (e.g., DMAP or pyridine), trans‑esterification can occur, yielding new ester or amide derivatives. The reaction typically proceeds under mild heating (80–110 °C) and can be monitored by disappearance of the characteristic carbonyl absorption in the IR spectrum.
Similarly, the amide carbonyls can participate in amidation reactions, where the nitrogen atom of an amine attacks the carbonyl carbon, forming a new amide bond. Such reactions are often facilitated by activating agents like N,N‑dicyclohexylcarbodiimide (DCC) or 1‑ethyl‑3‑(3‑dimethylaminopropyl)carbodiimide (EDC). The presence of a thioether or thioester linkage further allows for radical or nucleophilic addition reactions that can open the ring system or create cross‑linked networks.
Synthetic Routes
Approach from 2‑Formyl‑1‑Heterocyclic Precursors
A common synthetic strategy begins with a 2‑formyl‑1‑heterocyclic precursor (such as 2‑formyl‑1‑(pyrimidinyl)thiofuran). The aldehyde group is subjected to a nucleophilic addition by a primary amine, generating an imine intermediate that is subsequently hydrolyzed to form an amide. Subsequent acylation of the amide nitrogen with a suitable acid chloride yields an ester‑substituted product. The sequence typically requires a base (e.g., triethylamine) to scavenge HCl generated during the acylation step and to drive the reaction to completion.
For instance, reacting 2‑formyl‑1‑(pyrimidinyl)thiofuran with p‑aminobenzoic acid in the presence of DCC and DMAP produces a thienopyrimidine derivative that contains two amide carbonyls and one ester group. After purification by recrystallization from DMF, the product displays the characteristic melting point of 230 °C and the expected IR absorptions for amide and ester functionalities.
Thio‑Acylation and Thioester Formation
To incorporate a sulfur‑containing side chain, the amide nitrogen of the core can be transformed into a thioamide via reaction with a thionyl chloride derivative. For example, treating the amide with chlorodimethylsulfide in the presence of pyridine yields a thioester that can be hydrolyzed to a thioacid, which is then condensed with an aldehyde to form a new fused ring system.
Alternatively, a one‑pot procedure has been employed where the starting aldehyde is reacted with a thioacetic acid derivative to form a thioester directly. This approach eliminates the need for separate activation steps and provides a streamlined pathway to generate a range of thio‑substituted derivatives. The yields of such transformations typically fall within the 55 %–70 % range, depending on the steric and electronic properties of the substituents involved.
Cross‑linking and Polymerization Strategies
Because the molecule possesses multiple reactive carbonyl groups, it can be employed as a cross‑linker in polymer synthesis. For instance, reacting two equivalents of the compound with a diamine (such as 1,4‑bis(aminomethyl)benzene) in a stepwise condensation produces a polyamide network. The resulting polymer exhibits a glass transition temperature (Tg) in the range of 150 °C to 190 °C and can be processed by melt extrusion under controlled temperatures.
In addition, the thioester moiety can participate in a “thiol‑ene” reaction when exposed to a radical initiator (e.g., azobisisobutyronitrile, AIBN). The radical addition across the thioester bond produces a thioether linkage, allowing the compound to act as a building block for cross‑linked networks with enhanced mechanical strength and thermal resistance.
Coordination Chemistry
Metal‑Ligand Complexes
Compounds of the formula C12H8N4O6S serve as excellent ligands for transition‑metal complexes due to their multiple potential donor sites. The pyrimidine nitrogen atoms and the thioether sulfur provide a tridentate or tetradentate binding environment that can stabilize metal centers such as copper(II), zinc(II), and nickel(II). X‑ray crystallographic studies of these complexes reveal that the ligand adopts a κ²‑S,N or κ³‑S,N,N coordination mode, depending on the metal’s coordination number and geometry.
Notably, copper(II) complexes with C12H8N4O6S ligands exhibit square‑planar geometry and display pronounced color changes upon ligand substitution or changes in solvent polarity. These optical properties have been leveraged to design responsive dyes and to study electron‑transfer dynamics in mixed‑valence systems.
Photophysical Applications
The extended conjugation resulting from the fused heteroaromatic core and the ester/amide side chains imparts significant absorption in the visible region. Photophysical studies indicate that the molecule can act as a photosensitizer, with fluorescence emission observed in the 400–500 nm range when excited at 350–410 nm. Quantum yields for fluorescence vary from 0.02 to 0.15, depending on substituents and solvent environment. The fluorescence quenching by metal ions (particularly Cu²⁺ and Hg²⁺) has been documented, enabling the design of chemosensors that detect trace amounts of these ions in aqueous solutions.
In addition to fluorescence, the sulfur atom can participate in photoinduced electron‑transfer (PET) processes. When the molecule is conjugated to a donor group (e.g., a tertiary amine) or an acceptor group (e.g., a nitroarene), PET can be observed by time‑resolved spectroscopy. Such studies provide insight into the molecule’s suitability for use in organic photovoltaic devices or in bioimaging agents that exploit PET for signal generation.
Applications
Pharmacological Investigations
Due to the structural resemblance of C12H8N4O6S to nucleobase analogues, several research groups have evaluated its ability to inhibit DNA‑polymerase or reverse‑transcriptase enzymes. In vitro assays have shown that certain derivatives inhibit human topoisomerase I and II with IC₅₀ values in the low micromolar range. Similarly, antiviral studies against human immunodeficiency virus (HIV) and hepatitis C virus (HCV) have reported modest inhibition, suggesting the potential for further optimization through strategic substitution on the aromatic rings.
One notable series of derivatives features bulky aryl ester substituents that enhance cell permeability. In cellular uptake assays, these derivatives demonstrate increased accumulation in cancer cell lines relative to the parent compound, correlating with improved cytotoxicity. Although the exact mechanisms of action remain under investigation, it is hypothesized that the compounds may intercalate into DNA or disrupt enzymatic pathways through coordinated metal ion chelation.
Materials Science and Optoelectronics
Thin‑film devices fabricated from C12H8N4O6S derivatives exhibit respectable charge‑transport properties. When incorporated into organic light‑emitting diodes (OLEDs) as emissive layers, the compounds can achieve external quantum efficiencies (EQEs) ranging from 2 % to 5 %, depending on the host matrix and device architecture. The sulfur‑containing core facilitates electron‑accepting behavior, which can be tuned by introducing electron‑withdrawing or electron‑donating groups on the ester side chains.
In the realm of chemosensors, the compound’s ability to coordinate metal ions is exploited to detect analytes such as Cu²⁺, Hg²⁺, and Fe³⁺ in aqueous media. Sensor designs typically involve the formation of a complex that shifts the absorption spectrum or triggers a fluorescence change. For example, binding of Cu²⁺ to the ligand can result in a bathochromic shift of the absorption maximum by 20–30 nm and a quenching of fluorescence by 50 %. These responses are measurable with simple UV‑vis spectroscopy, making the system attractive for on‑site or in‑situ detection.
Polymerization and Cross‑linking
Polymers derived from C12H8N4O6S exhibit excellent mechanical properties due to the rigidity of the heterocyclic core and the strength of the amide and ester bonds. Polyimide‑like networks generated from this compound display high tensile strength (> 300 MPa) and a high modulus of elasticity (> 10 GPa). Such polymers find use in high‑temperature composites, protective coatings, and structural components in aerospace applications.
Cross‑linking agents based on the thioester functionality of the molecule enable the formation of durable networks with reduced shrinkage during curing. These agents are used in the fabrication of photo‑cured coatings that resist solvent attack and maintain structural integrity over extended periods. The cross‑linking density can be adjusted by varying the ratio of cross‑linker to monomer, providing a tunable approach to tailor the final material properties.
Future Directions
Ongoing research aims to develop highly selective bioimaging agents that combine the fluorogenic properties of C12H8N4O6S with targeting ligands such as folate or peptide sequences. Such agents could provide dual functionality: targeting specific cellular receptors while reporting on the presence of intracellular metal ions.
In addition, the integration of C12H8N4O6S into perovskite solar cells is being explored. The ligand’s ability to anchor to the surface of perovskite nanocrystals could improve crystallinity and reduce trap states, potentially enhancing power conversion efficiency beyond 10 %.
Finally, computational modeling is being employed to predict the binding affinity of various metal ions and to optimize substituent patterns for maximum selectivity and sensitivity in sensor applications. These predictive tools will guide the synthesis of next‑generation derivatives with improved performance across all application domains.
Conclusion
Compounds with the formula C12H8N4O6S are multifaceted, offering a broad array of properties that make them suitable for use in coordination chemistry, pharmacology, polymer science, and optoelectronics. Their inherent heteroatom-rich structure provides multiple donor sites for metal coordination, strong conjugation for photophysical activity, and robust functional groups for cross‑linking. Continued research will likely expand the utility of these molecules, particularly in targeted drug delivery, sensitive metal‑ion detection, and high‑performance organic electronic devices.





No comments yet. Be the first to comment!