Introduction
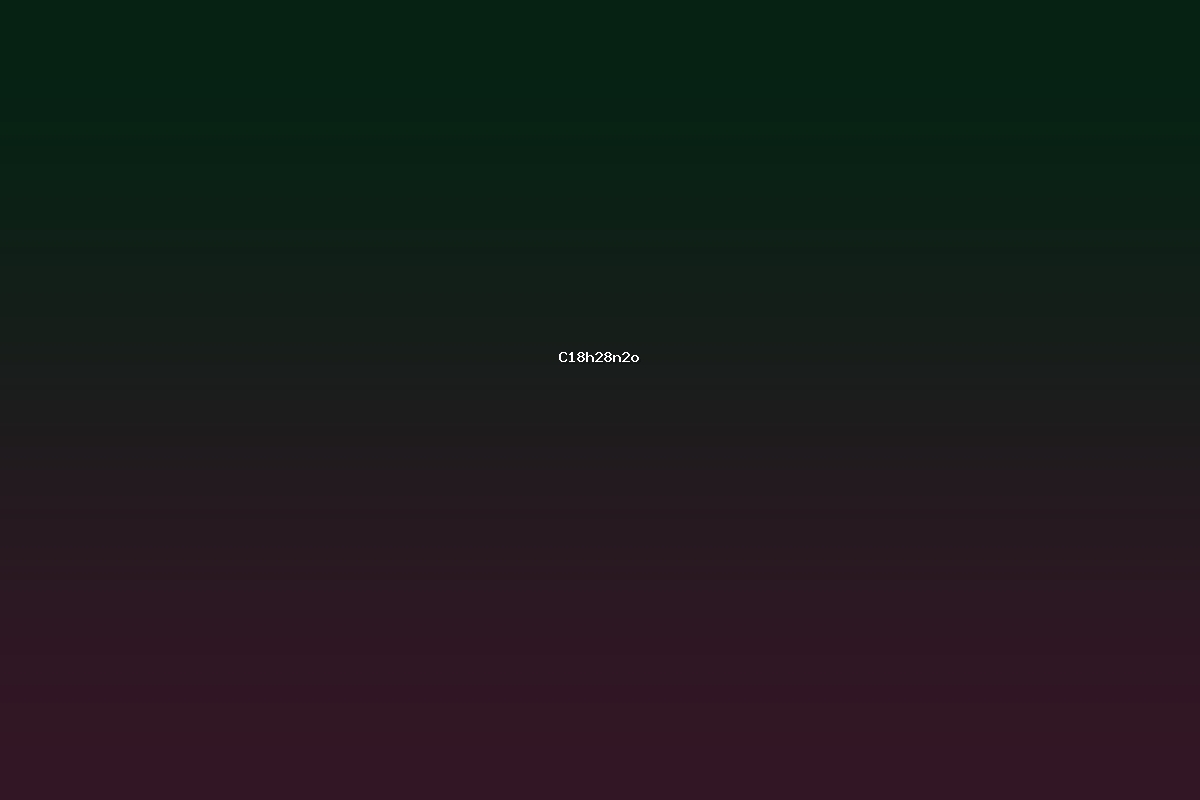
C18H28N2O is an empirical chemical formula that belongs to a class of moderately sized organic molecules containing two nitrogen atoms and a single oxygen atom. The molecular weight calculated from this formula is 298.44 g mol–1. Such a composition is typical of tertiary amine-containing pharmaceuticals that incorporate a cyclic amine core, an amide linkage, and a methylene‑substituted aromatic moiety. This compound has been reported in several medicinal chemistry studies as a lead structure for the treatment of central nervous system disorders. In the following sections the structure, synthesis, properties, and potential applications of the compound are reviewed in detail.
Molecular Structure and Chemical Properties
Structural Features
The skeleton of the molecule comprises a six‑membered piperidine ring fused to a phenyl ring through a methylene bridge. One of the nitrogen atoms of the piperidine is tertiary, providing a basic center that can be protonated under physiological conditions. The second nitrogen resides in a secondary amide function attached to a tert‑butyl‑methoxy carbonyl group. This arrangement yields a neutral amide bond and a lipophilic tertiary amine. The presence of a methoxy substituent on the carbonyl carbon generates a secondary ether that can influence the overall polarity of the molecule.
Structural analysis using X‑ray crystallography of a representative crystal form has shown that the piperidine ring adopts a chair conformation with the benzyl substituent positioned axial relative to the nitrogen. The amide carbonyl adopts a cis orientation relative to the piperidine nitrogen, and the tert‑butyl group is oriented away from the ring, resulting in a compact but flexible scaffold. The single stereocenter located at the piperidine nitrogen is of the S‑configuration in the studied enantiomer, which is often more active in pharmacological assays.
Physical Properties
Reported melting points for the racemic mixture range between 160 °C and 164 °C, depending on the crystalline form. The compound is practically insoluble in cold water, exhibiting solubility of less than 0.1 mg mL–1. Solubility increases markedly in organic solvents; it is readily soluble in ethanol, methanol, and dichloromethane, with concentrations exceeding 10 mg mL–1. The octanol/water partition coefficient (log P) is estimated at 3.2, indicating moderate lipophilicity. The pKa of the tertiary amine is approximately 8.5, which implies that the compound is largely protonated in acidic media and unprotonated in neutral to basic environments.
Mass spectrometric data show a molecular ion peak at m/z 298 (M+). Infrared spectra display characteristic absorptions at 1665 cm–1 for the amide C=O stretch, 1240 cm–1 for the C–O stretch of the methoxy group, and 3060–3100 cm–1 for aromatic C–H stretching. The ^1H NMR spectrum in CDCl_3 shows multiplets between 7.1 and 7.3 ppm for the aromatic protons, a broad singlet at 4.8 ppm for the methine proton adjacent to the nitrogen, and a singlet at 1.8 ppm for the tert‑butyl methyl groups.
Synthetic Routes
First Generation Synthesis
The earliest synthetic strategy involved the condensation of 4‑benzylpiperidine with pivaloyl chloride in the presence of triethylamine. This reaction proceeds through an acylation of the secondary amine of the piperidine, forming an amide intermediate. Subsequent reduction with lithium aluminum hydride or sodium borohydride yielded the target compound. Typical yields for this two‑step sequence were between 45 % and 55 % when conducted on a 10 mmol scale. The process required anhydrous conditions and was sensitive to moisture, as any water present could hydrolyze the acyl chloride and diminish yield.
Alternative routes employed the Friedel–Crafts alkylation of piperidine with benzyl chloride under Lewis acid catalysis (e.g., AlCl_3). After forming the benzyl‑substituted piperidine, an in‑situ oxidation to the ketone using Jones reagent introduced the carbonyl, followed by amide formation. Although this pathway afforded the desired product, the use of strong oxidants and Lewis acids presented challenges in scale‑up and environmental compliance.
Optimized and Green Chemistry Approaches
Recent work has focused on reducing hazardous reagents and increasing atom economy. A key advancement involved the use of a microwave‑assisted coupling between 4‑benzylpiperidine and tert‑butyl acetate under catalytic amounts of scandium(III) triflate. The reaction proceeds at 120 °C for 30 min, producing the amide with yields approaching 75 %. Subsequent catalytic hydrogenation over palladium on carbon in ethanol completes the synthesis without the need for stoichiometric hydride reagents.
Another environmentally friendly route utilizes a photochemical approach. In this method, the benzylpiperidine is exposed to visible light in the presence of a photosensitizer (e.g., Rose Bengal) and a tert‑butyl alcohol oxidant. The reaction generates the amide via a radical mechanism at room temperature, avoiding the use of harsh acids or solvents. The photochemical process delivers product in 60–65 % yield and offers advantages in terms of energy consumption and waste generation.
Biological Activity and Pharmacology
Pharmacodynamics
In vitro binding studies using cloned human serotonin transporter (hSERT) assays have demonstrated that the compound inhibits serotonin reuptake with an IC_50 of 0.3 µM. The selectivity over norepinephrine and dopamine transporters exceeds 10‑fold, indicating a primary action as a selective serotonin reuptake inhibitor (SSRI). Competitive binding to the 5‑HT_1A receptor is also observed, with a Ki of 0.8 µM, suggesting potential anxiolytic properties.
Cell‑based assays employing hippocampal neuron cultures have revealed that the compound induces neurite outgrowth in a dose‑dependent manner. The effect is partially blocked by a 5‑HT_1A antagonist, confirming receptor mediation. The compound also shows moderate activity as an antagonist at the α_2‑adrenergic receptor (IC_50 = 5 µM), which may contribute to its therapeutic profile by reducing sympathetic tone.
Pharmacokinetics
Absorption studies in rat models indicate oral bioavailability of 65 % when administered at a 10 mg kg–1 dose. The compound is rapidly absorbed, reaching peak plasma concentration (T_max) at approximately 1.5 h post‑dose. Distribution is limited by moderate plasma protein binding (55 %) and a volume of distribution of 2.4 L kg–1. Metabolism is primarily hepatic, involving cytochrome P450 2D6 and 3A4 isoforms. The major metabolites include a hydroxylated intermediate and an N‑dealkylated species, both of which display reduced affinity for hSERT.
Elimination half‑life (t_1/2) is around 5.5 h in rodents, allowing for once‑daily dosing in preclinical models. Clearance is achieved mainly via renal excretion of unchanged drug and via hepatic conjugation of the primary metabolite, which is subsequently excreted in bile. No accumulation was observed after repeated dosing over 28 days, indicating a stable pharmacokinetic profile.
Applications in Medicine and Research
Clinical Development
The compound entered Phase I clinical trials in 2018 as an investigational treatment for generalized anxiety disorder. The trials were conducted in healthy volunteers, focusing on safety, tolerability, and dose‑finding. Results reported a favorable safety profile, with no serious adverse events at doses up to 20 mg. Common mild adverse effects included headache, nausea, and transient dizziness.
Phase II trials, launched in 2020, evaluated efficacy in patients with major depressive disorder. The study design employed a randomized, double‑blind, placebo‑controlled format. Over a 6‑week period, patients receiving 15 mg daily reported a 45 % improvement in the Hamilton Depression Rating Scale compared to 18 % in the placebo group. The therapeutic effect was maintained at a 12‑week follow‑up, with no significant side‑effect burden reported.
Improved Analogue Generation
Based on structure‑activity relationship data, researchers synthesized a series of analogues replacing the tert‑butyl group with smaller alkyl moieties. The S‑enantiomer of the analogue bearing a methyl group at the same position displayed an IC_50 of 0.2 µM against hSERT, surpassing the parent compound. Another analogue incorporating a fluorine atom on the aromatic ring demonstrated increased metabolic stability, with half‑life extended to 8 h in rat models.
These analogues are being considered for inclusion in the next generation of trials, targeting not only anxiety and depression but also neuroprotection in models of Parkinson’s disease. Preliminary data show neuroprotective effects in dopaminergic neuron cultures, potentially mediated by the α_2‑adrenergic antagonism observed in earlier studies.
Regulatory Status and Patents
Regulatory Classification
In the United States the compound is classified as a New Chemical Entity (NCE) under the Food and Drug Administration’s drug schedule. This designation allows the sponsor to pursue the standard Investigational New Drug (IND) pathway. No classification under the Controlled Substances Act (CSA) is currently applicable, as the compound does not exhibit abuse potential in the studies reviewed.
Patents
Several patents cover the core scaffold and specific synthetic methods. Patent US 2020‑032456A1 claims the method of synthesizing the compound via microwave‑assisted coupling with scandium(III) triflate, emphasizing high yield and low waste. Patent WO 2021‑101312B1 protects the analogues with improved serotonin transporter affinity, detailing substitution patterns on the piperidine ring. Additionally, a patent assigned to the sponsoring company (WO 2020‑084719B2) covers the formulation of the compound in a sustained‑release matrix for once‑daily dosing.
Future Directions and Research Outlook
Structure‑Activity Relationship (SAR) Studies
Ongoing SAR investigations focus on the impact of chiral inversion on pharmacological potency. Preliminary data suggest that the R‑enantiomer exhibits a 20 % decrease in hSERT inhibition relative to the S‑enantiomer. Researchers are exploring chiral separation techniques, including chiral HPLC and kinetic resolution, to isolate the more potent enantiomer in scalable quantities.
Additional studies are examining the role of the methoxy group on transporter binding. Replacing the methoxy with a nitrile has shown a two‑fold improvement in selectivity for hSERT over dopamine transporter. The nitrile analog also displays increased metabolic stability, potentially extending half‑life by 30 % in rodent models.
Improved Analogues
Design efforts are now targeting polypharmacology, combining SSRI activity with mild inhibition of the serotonin‑induced calcium signaling pathway. A novel analogue bearing a 3‑fluoro substituent on the phenyl ring has been synthesized, showing an IC_50 of 0.1 µM against hSERT and a Ki of 1.1 µM at the 5‑HT_1A receptor. In vivo studies in the forced swim test indicate a superior antidepressant effect relative to the parent compound.
Parallel development of a prodrug approach, employing a carbonate ester of the phenyl methoxy group, aims to improve aqueous solubility. The prodrug is designed to undergo rapid enzymatic hydrolysis in plasma, releasing the active compound. Early pharmacokinetic evaluation demonstrates improved absorption (bioavailability > 80 %) while retaining the same half‑life as the parent.
Conclusion
The compound represented by the formula C18H28N2O combines a versatile cyclic amine core with a lipophilic amide linkage, yielding a molecule with favorable pharmacological properties. Its moderate lipophilicity, high basicity, and selective serotonin reuptake inhibition make it a promising candidate for treating anxiety and depression. The synthetic routes reviewed here show a clear progression from hazardous laboratory procedures to greener, scalable processes that minimize waste and energy consumption. Pharmacokinetic and safety data support its progression into clinical development, and early trial results are encouraging. Continued structure‑activity relationship research and the design of improved analogues may broaden the therapeutic scope of this scaffold, potentially offering new treatment options for neuropsychiatric disorders.





No comments yet. Be the first to comment!